IFOM research programs
Discover our research programs
RAS Oncogenic Signaling and Therapeutic Resistance
Principal Investigator
Chiara Ambrogio
Functional Proteomics
Principal Investigator
Angela Bachi
Genomics of Cancer and Targeted Therapies
Principal Investigator
Alberto Bardelli
DNA Repair
Principal Investigator
Dana Branzei
Artificial Intelligence & Systems Biology
Principal Investigator
Francesca Buffa
Genetics of B Cells and Lymphomas
Principal Investigator
Stefano Casola
RNA regulatory networks in translational oncology
Principal Investigator
Matteo Cereda

Quantitative Biology of Cell Division
Principal Investigator
Andrea Ciliberto
Statistical physics of cells and genomes
Principal Investigator
Marco Cosentino Lagomarsino

DNA Metabolism
Principal Investigator
Vincenzo Costanzo
DNA Damage Response and Cellular Senescence
Principal Investigator
Fabrizio d'Adda di Fagagna
Signalling, tumor environment and cell metabolism
Principal Investigator
Giannino Del Sal

Replication Stress Response
Principal Investigator
Ylli Doksani
Computational genomics
Principal Investigator
Francesco Ferrari
Genome Integrity
Principal Investigator
Marco Foiani

Mechano-Oncology
Principal Investigator
Nils Gauthier
Cell Plasticity & Aging
Principal Investigator
Marta Kovatcheva
Chromosome Instabilities
Principal Investigator
Makoto Hayashi
Functional Genomics of Cancer Immunity
Principal Investigator
Giuseppe Leuzzi
Precision Oncology
Principal Investigator
Silvia Marsoni
Molecular Oncology and Immunology
Principal Investigator
Massimo Pagani
Tissue biology and tumorigenesis
Principal Investigator
Stefano Piccolo
Molecular Machines in Signalling Pathways
Principal Investigator
Simona Polo
Tumor Spatial Biology
Principal Investigator
Denis Schapiro
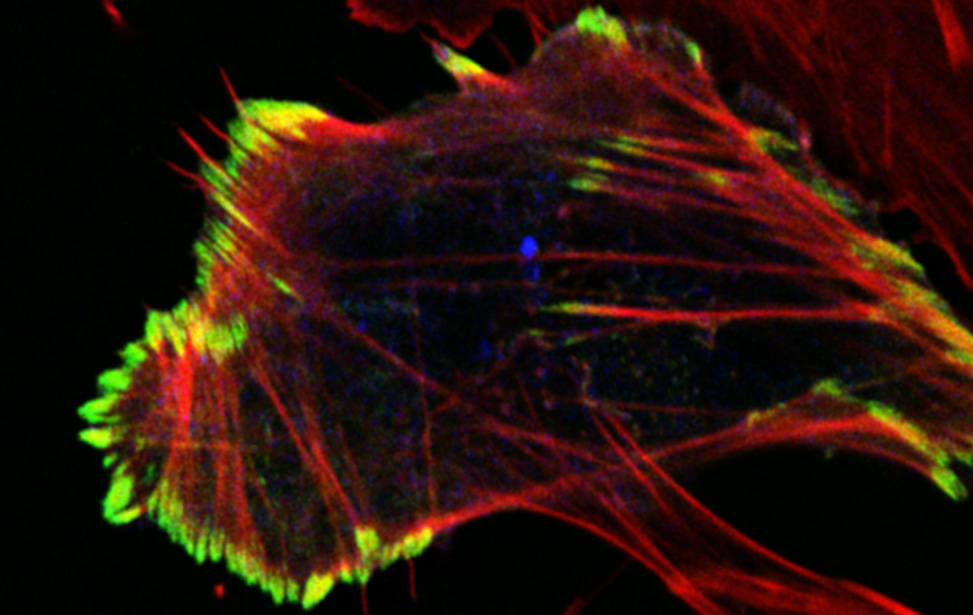
Mechanisms of Tumor Cell Migration
Principal Investigator
Giorgio Scita

Advanced Pathology Laboratory
Principal Investigator
Claudio Tripodo
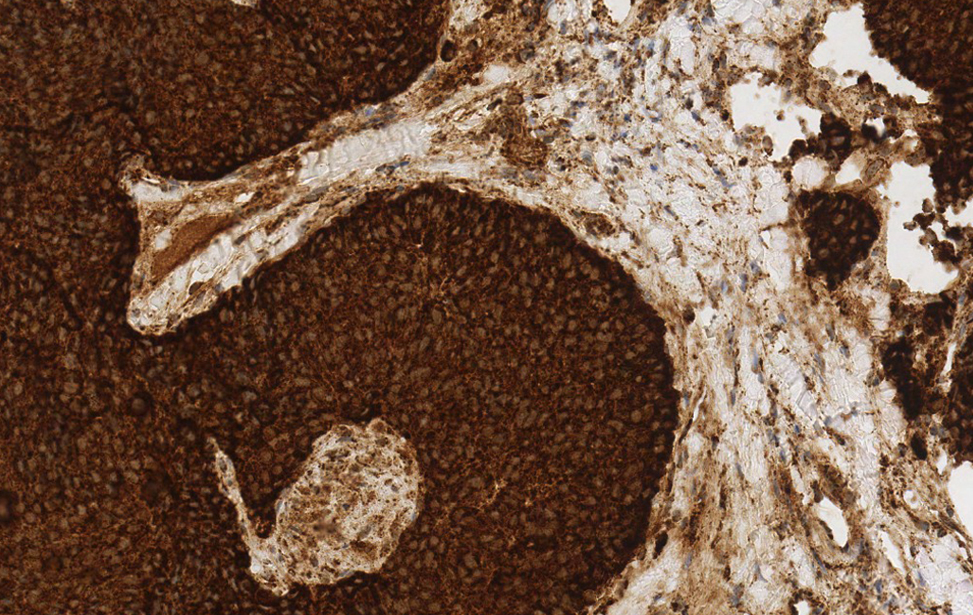
Metabolic Reprogramming in Solid Tumors
Principal Investigator
Claudio Vernieri
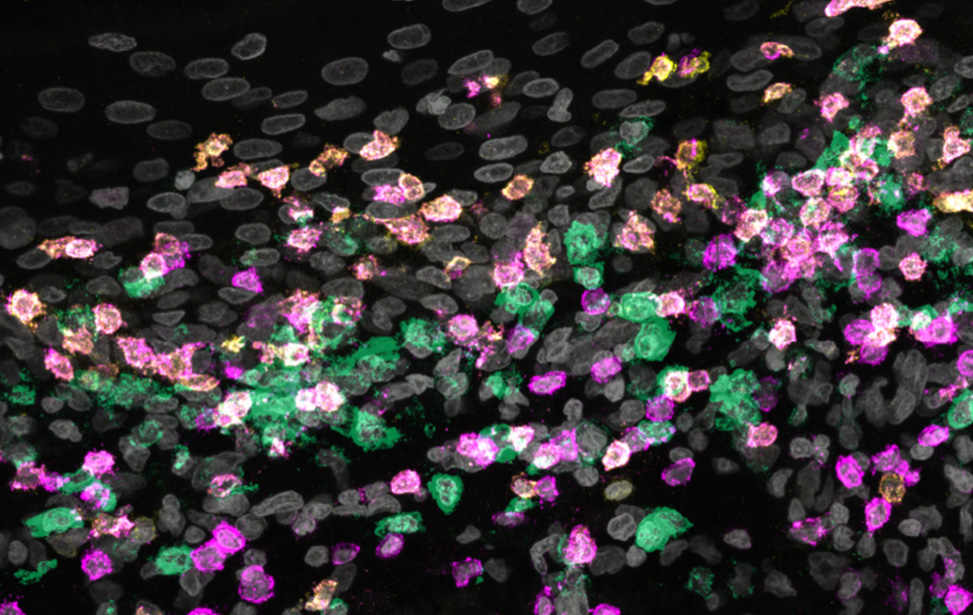
Tumor Microenvironment and Immunotherapy
Principal Investigator
Beatrice Zitti