Computational genomics
Background
Our research group name "computational genomics" reflects our area of technical expertise in computational biology and genomics, but our research interests and biological expertise are focused on chromatin three-dimensional (3D) architecture.
The 3D organization of chromatin within the cell nucleus is crucial for regulating genome functionality. The knowledge of its role has greatly advanced over the few years thanks to the development of novel imaging, molecular biology, computational data analysis and modelling techniques.
My research group is especially interested in understanding how chromatin 3D architecture is connected to epigenetic and transcriptional regulation of genome functionality, and how this regulatory axis ultimately affects cell identity definition and homeostasis in physiological and pathological conditions, with a special focus on cancer.
We can distinguish different levels of 3D structural organization of chromatin, that correspond to different molecular mechanisms acting on chromatin and the genome. Thus, chromatin architecture is studied at different levels of resolution:
On a large scale, we are interested in mechanisms for the coordinated control of large chromatin domains in physiological and disease conditions. These involve, among the others, the compartmentalization of the genome in distinct structural domains, such as Topologically Associating Domains (TADs), or Lamina Associated Domains (LADs), as well as the functional and biochemical distinction between heterochromatin and euchromatin regions. We investigate their functional and structural reorganization dynamics in cancer.
On a finer scale, instead, we study how chromatin 3D folding comes into play to control the physical association of distal regulatory elements (enhancers) and their target genes. In this context we combine biological knowledge, experimental data and computational approaches to reconstruct the broader regulatory network encompassing genes and their non-coding regulatory elements (promoters and enhancers). We use this complex network or physical (in the 3D space) and regulatory (epigenetics and transcriptional) connections as a lens to understand and characterize the multivariate effect of epigenetic or genetic alterations in cancer.
Ongoing research projects
Our group includes both experimental (wet lab) and computational (dry lab) expertise, that we apply to dissect molecular mechanisms and biological processes involved in cancer.
- Heterochromatin dynamics in cancer cell plasticity.
We recently developed a unique set of experimental techniques (SAMMY-seq and 4f-SAMMy-seq) to study chromatin architecture and especially heterochromatin dynamics with unprecedented versatility and sensitivity (Sebestyén et al., Nature Communications 2020; Lucini et al., Nucleic Acids Res. 2024). These techniques allow studying chromatin compartments rearrangements in a variety of experimental settings (e.g. small biological samples such as needle biopsies) that would be challenging for other state of the art methods for chromatin 3D architecture analysis. In ongoing research projects, we are leveraging these techniques i) to identify the molecular determinants of signatures of epigenetic plasticity in cancer samples, and ii) to dissect the molecular mechanisms connecting epigenetic requiring, phenotypic plasticity and response to tumor microenvironment signals. - Altered enhancer-genes regulatory networks in cancer.
Distal non-coding regulatory elements (enhancers) get in close physical proximity of their target gene(s) thanks to the three-dimensional folding of chromatin. We recently developed computational and statistical methods to integrate chromatin 3D architecture data in the reconstruction of enhancer-gene regulatory interactions (Salviato et al., Nucleic Acids Research 2021) and applied them to characterize the functional targets of non-coding regulatory mutations in cancer patients (Hariprakashet al., Cancer Research 2024). In ongoing research projects, we are i) further expanding these computational methods to leverage single cell genomics data to identify transcriptional and epigenetics regulatory modules activated in heterogenous or rare cell sub-populations; ii) to applying this type of integrated epigenomic data analysis to characterize epigenetic rewiring in cancer cells. - Chromatin architecture data analysis methods.
Our research work is driven by biological on mechanisms of epigenetics and transcriptional regulation in cancer. However, we often find that established computational genomics methods do not allow us to address biological problems we are investigating. Thus we actively work also on novel computational biology methods for the analysis of functional genomics data, in particular for epigenomics and 3D chromatin architecture data (Pal et al., Nature Communications 2019; Pal et al., Bioinformatics 2019; Forcato et al., Nature Methods 2017).
29-05-2024 rel. 03

Photogallery
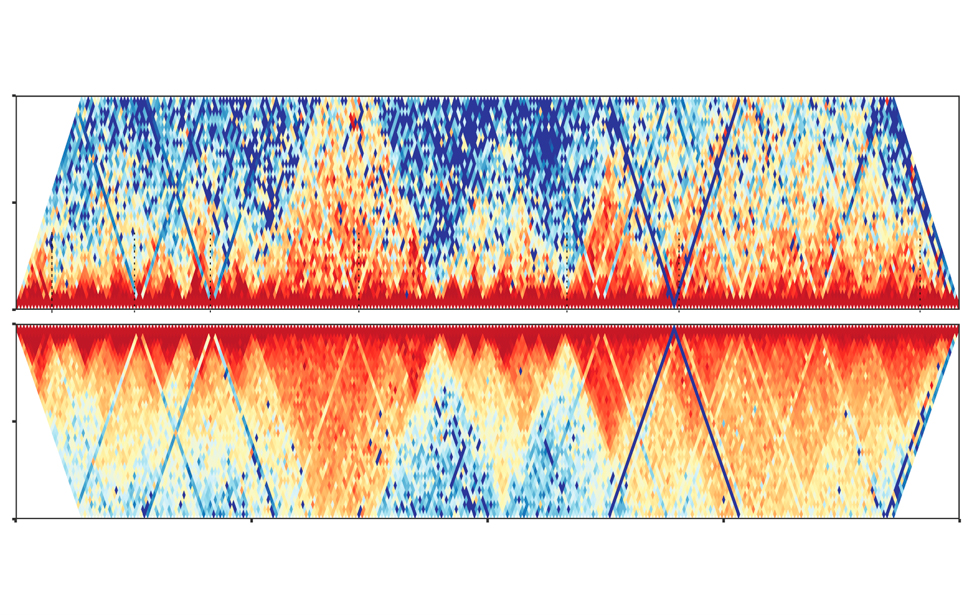 Chromatin 3D architecture contact matrices
Chromatin 3D architecture contact matrices
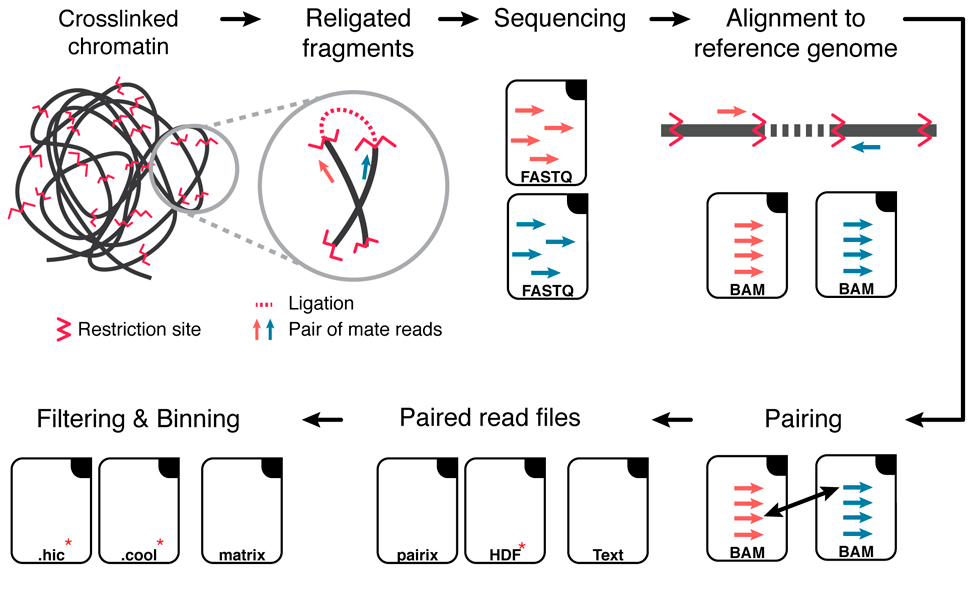 Hi-C Data analysis workflow
Hi-C Data analysis workflow
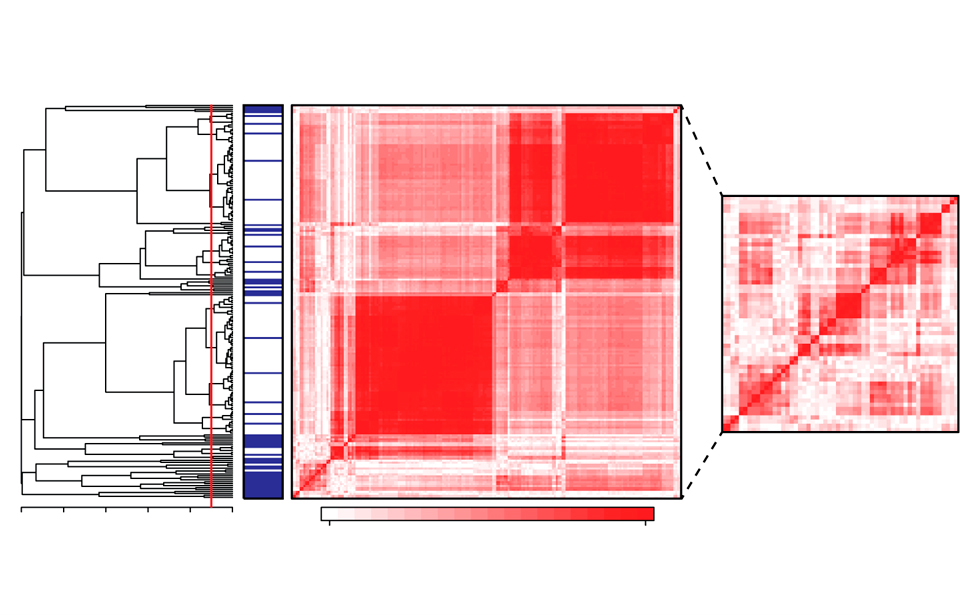 Features selection for machine learning on epigenomics data
Features selection for machine learning on epigenomics data
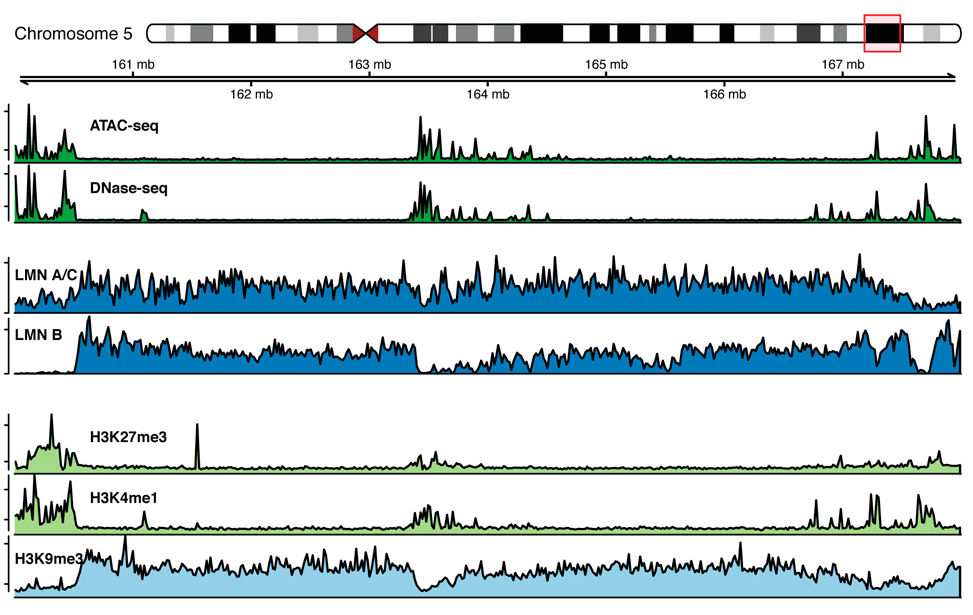 Chromatin marks profiles around lamina associated domains
Chromatin marks profiles around lamina associated domains
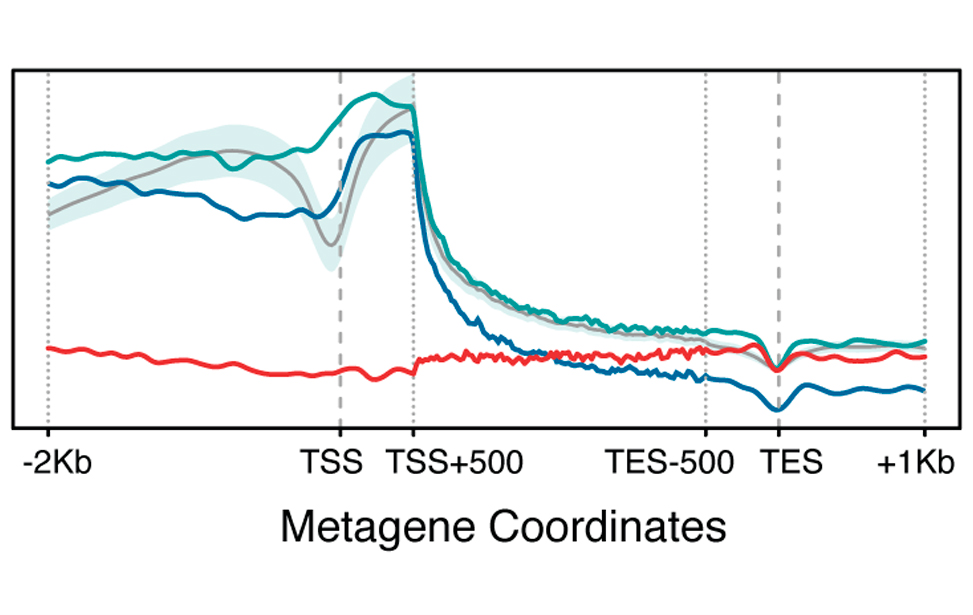 Epigenetic marks profiles
Epigenetic marks profiles