DNA damage Response and Cellular Senescence
Research project
DNA damage response (DDR) and cellular senescence
We study the physiological consequences of DNA damage at the cellular and organismal level mainly in mammals. Nuclear DNA damage triggers a signaling and effector pathway known as the DNA damage response (DDR) that coordinates cell-cycle arrest (checkpoint functions) and DNA repair. Persistent DDR signaling establishes cellular senescence, a condition in which cells remain alive but permanently unable to further proliferate (Campisi and d'Adda di Fagagna, Nature Reviews Molecular Cell Biology 2007; d'Adda di Fagagna, Nature 2003).
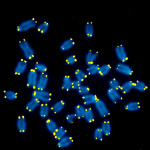
Figure 1. Mouse metaphase chromosomal spreads. DNA is blue, telomeric TTAGGG repeats are in yellow.
Previous achievements
Cellular senescence is crucial in the study of both ageing and cancer. We have worked on both of these fundamental processes.
Most normal human cells can undergo only a limited number of cells divisions before reaching the so-called "Hayflick limit" and entering the non-proliferative condition known as replicative cellular senescence. This arrest is dictated by telomeres, the tips of linear chromosomes. We have demonstrated that replicative cellular senescence is the outcome of the direct recognition of critically short telomeres by the DDR machinery that senses them as DNA lesions and enforce a permanent proliferative arrest. This has firmly placed the DDR as a causative mechanism of cellular senescence (d'Adda di Fagagna, Nature 2003). More recently, we demonstrated that also non proliferating cells accumulate damage at telomeres in the absence of telomere attrition, thus pointing to telomeric DNA damage and DDR activation as a common element of cell aging in both proliferating and non proliferating tissues (Fumagalli, Nature Cell Biology 2012; Rossiello, Current Opin Genet Dev 2014).

Figure 2. Fabrizio d'Adda di Fagagna next to Leonard Hayflick.
Oncogene activation in normal human cells is not sufficient to transform them into cancerous cells. Rather, it leads to oncogene-induced cellular senescence (OIS). We have demonstrated that oncogene activation is intrinsically a genotoxic event due to altered DNA replication and that the consequent DDR activation is the causative molecular mechanism responsible for senescence establishment. Consistent with this, we have observed DDR activation in human tumor samples (Di Micco, Nature 2006; Nuciforo, Carcinogenesis 2007).
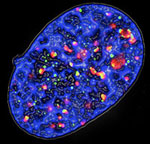
Figure 3. Fluorescence microscopy image of a senescent cell expressing oncogenic Ras: the phosphorylated form of histone H2AX (γ-H2AX) is stained in green and the DNA damage checkpoint protein 53BP1 is in red, DNA is in blue.
Cellular senescence is associated with the secretion of a complex mix of cytokines. We provided supporting evidence that some interleukins play a senescence-enforcing role by increasing DDR activation, thus establishing a feedback loop between DDR signaling and inflammation (Acosta, Cell 2008; Fumagalli, Nature Cell Biology 2009).
Several oncogenes are known to induce increased reactive oxygen species (ROS) levels responsible for DDR activation and senescence establishment. We identified a cellular pathway (?) that leads to ROS accumulation upon oncogene activation and demonstrated that ROS are mitogenic and cause DNA damage by fueling oncogene-induced aberrant DNA replication (Ogrunc, Cell Death & Differentiation 2014)
Cellular senescence can also be associated with a global increase of heterochromatin. We showed that heterochromatin is preferentially induced by oncogene-driven DNA replication stress and that it is not lost upon bypass of OIS in vitro and in vivo in human tumors. Rather, we have shown that heterochromatin acts by restraining the DDR induced by oncogenes, and that treating oncogene-expressing cells with heterochromatin-disrupting drugs, such as histone deacetylase inhibitors (HDACi), boosts DDR and selectively induces cell death by apoptosis in a DDR-dependent manner, thus providing one of the first examples of a senolytic treatment (Di Micco, Nature Cell Biology 2011).
We observed that the DDR in stem cells is differently regulated from somatic cells. In Caenorahbditis elegans germ stem cells the DDR is controlled by the NOTCH pathway in an evolutionary-conserved manner (Vermezovich, Nature Structural Molecular Biology 2015; Adamowicz, Cell Reports 2016; Adamowicz, DNA repair 2018). We also provided evidence that DNA damage to neural stem cells can induce cell differentiation both in cultured cells, as well as in mice (Schneider, Stem cells reports 2013).
Ongoing research
While cellular senescence in aging and cancer remains our interests, we recently made the surprising discovery that at sites of DNA damage non coding RNA are generated and processed, and we demonstrated that they are necessary for full DNA damage response (DDR) activation, including DNA damage signaling and repair (d'Adda di Fagagna, Trends in Cell Biology 2013; Michelini, Chemical Reviews 2018).
We discovered that RNA polymerase II is recruited by the MRE11-RAD50-NBS1 (MRN) complex at DNA double-strand breaks (DSB) where it synthesizes damage-induced long non coding RNA (dilncRNA) on both DNA strands. MRN supports transcription from DNA ends by melting them (Sharma, Cell Reports 2021) Such ncRNA can be processed by the endoribonucleases Drosha and Dicer to generate shorter ncRNA that we termed DNA damage response RNA (DDRNA) that can bind to unprocessed complementary dilncRNA and together they promote liquid-liquid phase separation (LLPS) of DDR factors at sites of DNA damage in the form of DDR foci (Francia, Nature 2012; Michelini, Nature Cell Biology 2017; Pessina Nature Cell Biology 2019). More recently we reported that the entire preinitiation complex (PIC) together with RNA polymerase II is recruited at DSB, thus in a manner similar to canonical transcriptional promoters (Pessina, Nature Cell Biology 2019). If DSB are resected, DNA:RNA hybrids can form and control DNA repair by homologous recombination (D'Alessandro, Nature Communications 2018). The pathways that leads to DDRNA formation can be modulated by small molecules (Gioia, Scientific reports 2019). Most excitingly, our findings provide a unique opportunity to inhibit DDR for the first time in a sequence-specific manner. Indeed, antisense oligonucleotides (ASO) against dilncRNA and DDRNA allow to inhibit DDR activation at individual damaged genomic sites, thus with unprecedented selectively (Michelini, Nature Cell Biology 2017).
Critically short and damaged telomeres are triggers of cellular senescence and likely contributors of aging and aging-related diseases. DilncRNA and DDRNA synthesis and accumulation occurs also at dysfunctional telomeres. We demonstrated that ASO against telomeric dilncRNA and DDRNA (tASO) allows selective DDR inhibition at telomeres, both in cultured cells and in vivo in mice (Rossiello, Nature Comunications 2017). By exploiting our ability to inhibit DDR selectively at telomeres with tASO, we improved cell proliferation, tissues homeostasis and extended lifespan in Hutchinson-Gilford Progeria Syndrome (HGPS) patients cells and in a mouse model of this accelerated aging disease (Aguado, Nature Communications 2019).
Presently we have a number of unpublished observations in additional mouse and fish models that demonstrate safety and efficacy of tASO in human conditions caused by telomere dysfunction. Targeting recurring sites of DNA damage accumulation in cancer cells is another opportunity we are exploring. As DNA damage is emerging as a potential causative mechanism for several forms of neurodegeneration, we are exploring this field leveraging on our scientific background.
Our approach is patented and we aim to take it to clinical settings for the benefit of patients.
07/06/2024 rel07

Photogallery
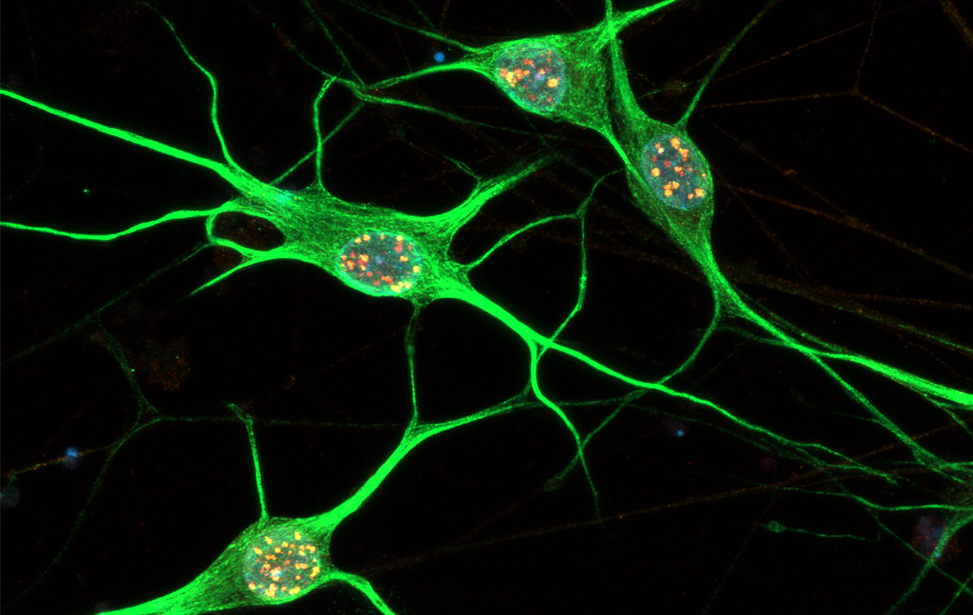 Immunostaining of human motor neurons upon IR treatment (2Gy). Merge signal of MAP2 (green), DAPI (blue), γH2AX (red) and pATM (magenta)
Immunostaining of human motor neurons upon IR treatment (2Gy). Merge signal of MAP2 (green), DAPI (blue), γH2AX (red) and pATM (magenta)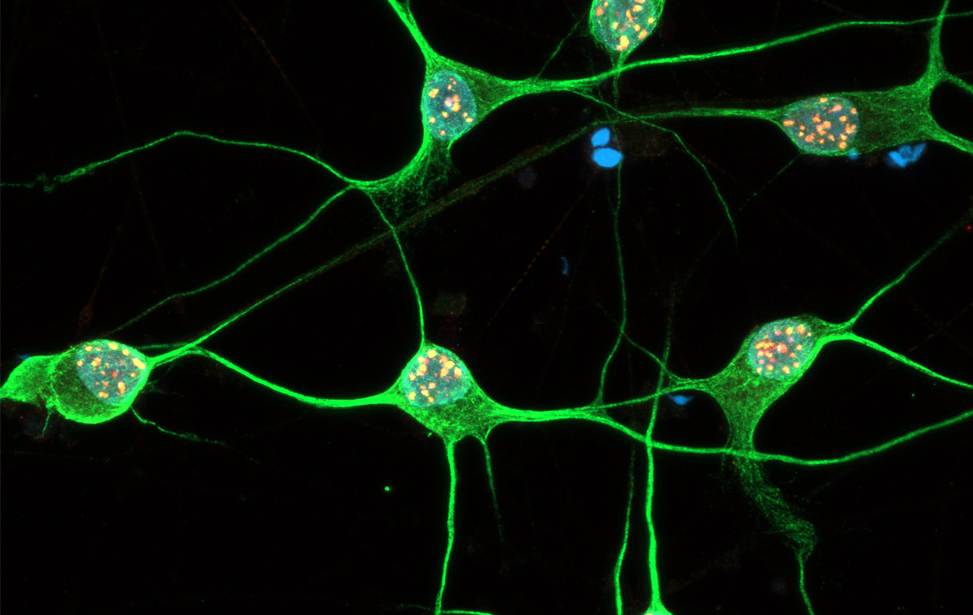 Immunostaining of human motor neurons upon IR treatment (2Gy). Merge signal of MAP2 (green), DAPI (blue), γH2AX (red) and pATM (magenta)
Immunostaining of human motor neurons upon IR treatment (2Gy). Merge signal of MAP2 (green), DAPI (blue), γH2AX (red) and pATM (magenta)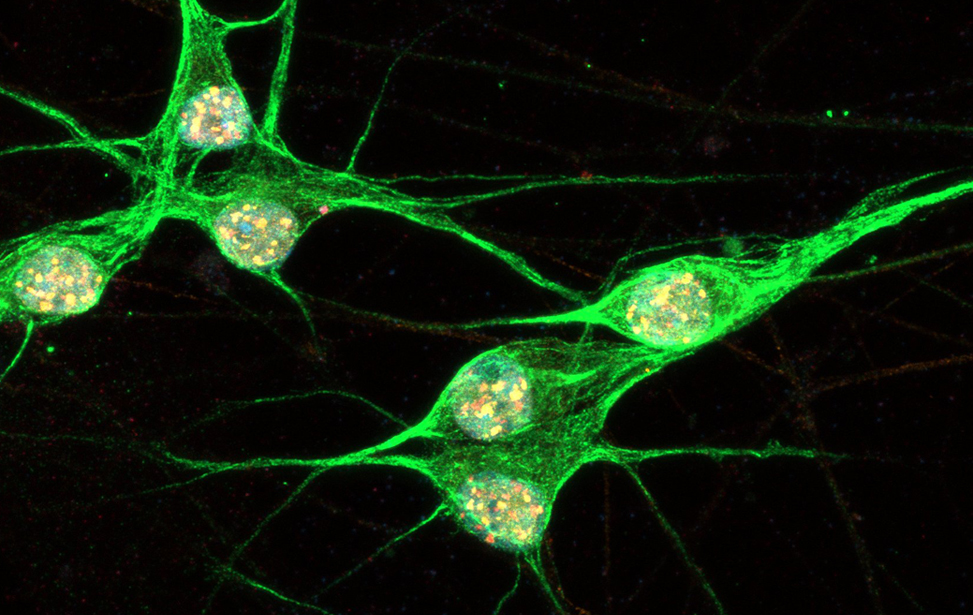 Immunostaining of human motor neurons upon IR treatment (2Gy). Merge signal of MAP2 (green), DAPI (blue), γH2AX (red) and pATM (magenta)
Immunostaining of human motor neurons upon IR treatment (2Gy). Merge signal of MAP2 (green), DAPI (blue), γH2AX (red) and pATM (magenta)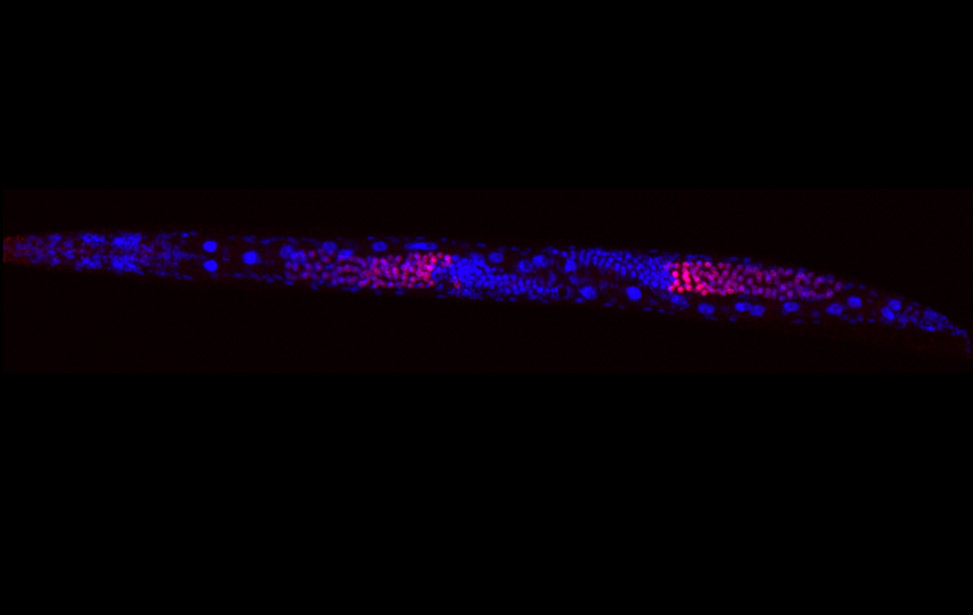 worm stq
worm stq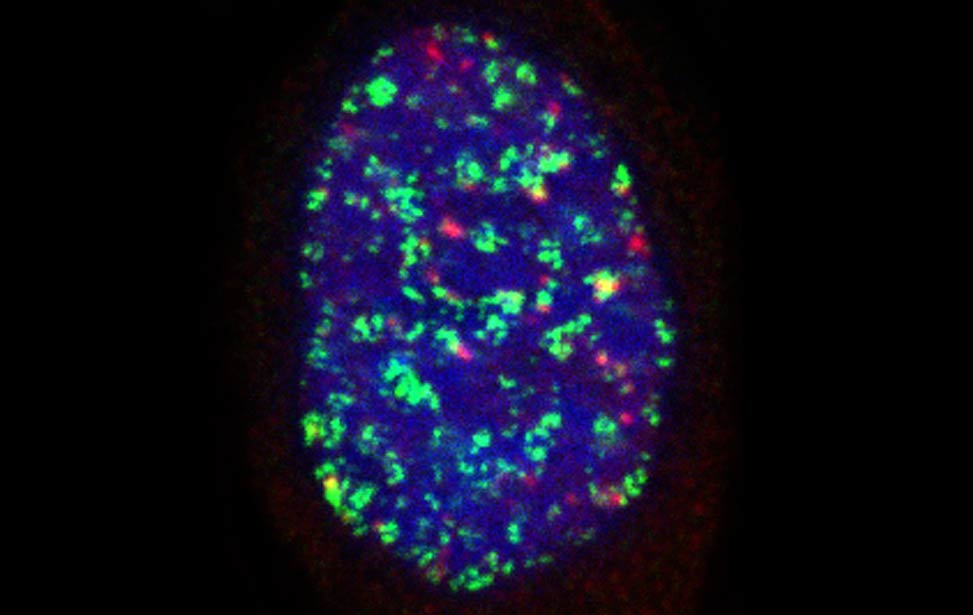 greenPCNA_red53BP1_rasD3
greenPCNA_red53BP1_rasD3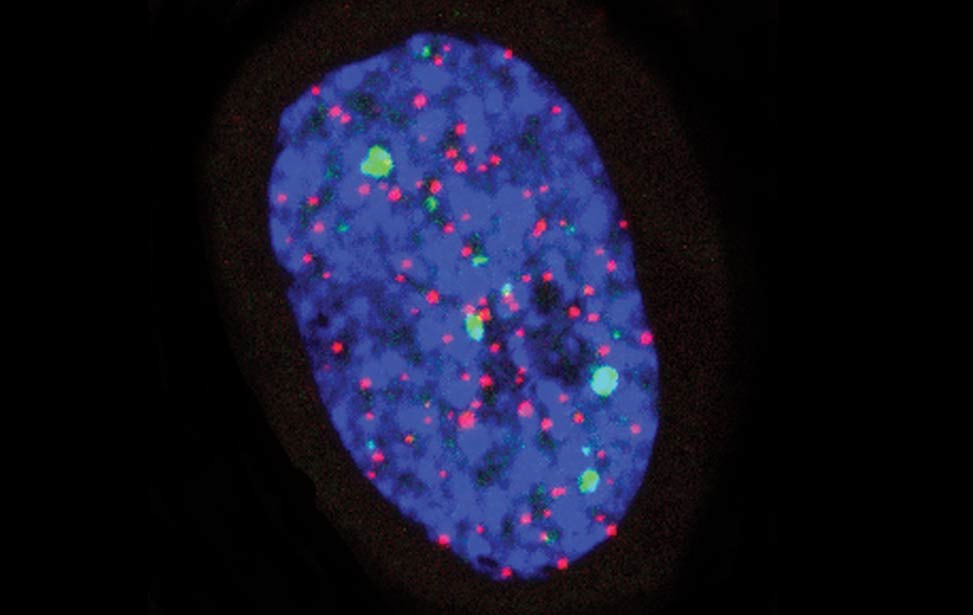 Fra - Senescent BJ DDR Telomeres
Fra - Senescent BJ DDR Telomeres FLA_NIH24
FLA_NIH24 Fra - Mouse brain telomeres
Fra - Mouse brain telomeres